
What is Congenital Adrenal Hyperplasia (CAH)?
A brief overview of CAH & it's forms
Congenital Adrenal Hyperplasia (CAH) is a family of inherited disorders affecting the adrenal glands
Notably, the most common form is 21-hydroxylase deficiency (21-OHD), is inherited in severe or mild forms. The severe form, called Classical CAH, is usually detected in the newborn period or in early childhood. The milder form, called Non-classical CAH (NCAH), may cause symptoms at any time from infancy through adulthood. NCAH is also a much more common disorder than Classical CAH.
Fortunately, one can manage CAH with medication and, with adequate care, allowing affected individuals to go on to live normal lives.
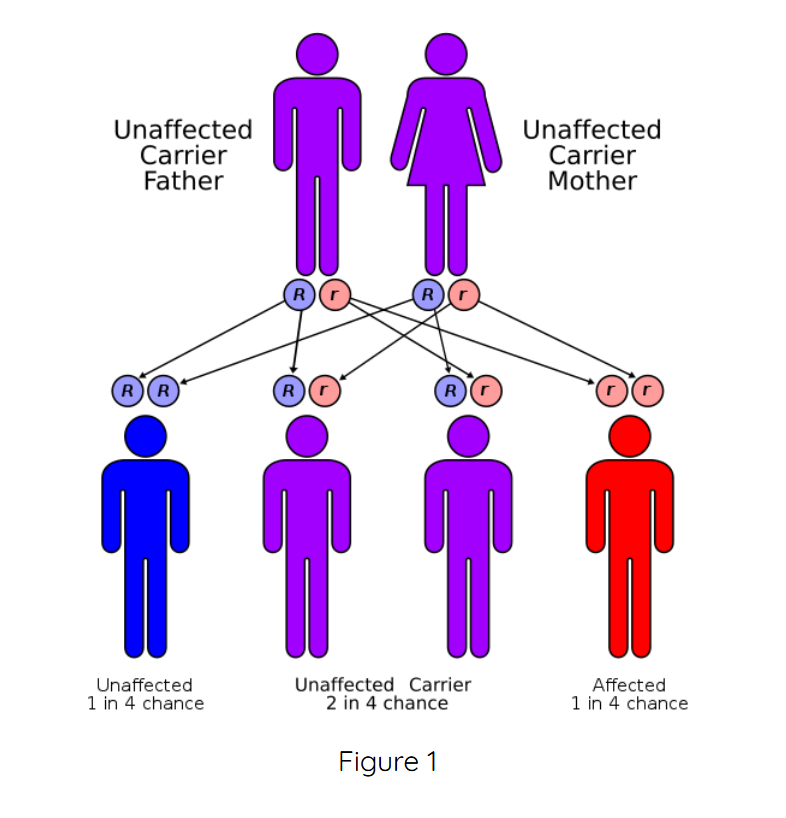
To summarize, CAH is an autosomal recessive genetic disorder. It affects males and females in equal numbers. However, for a child to be born with either form of CAH, both parents must carry a gene for the disorder. Scientists have also pinpointed the location of the group of genes that causes the most common forms of CAH to chromosome 6. Additionally, DNA testing is available for diagnosis of CAH and to detect carriers of the gene mutations.
These particular groups of genes contain instructions the adrenal glands (located on top of the kidneys) need in order to produce an enzyme called 21-hydroxylase. As a result, without this enzyme, the adrenal glands would thus be unable to produce cortisol, a hormone necessary for life.
Cortisol is a steroid produced by the adrenal glands that our bodies need to: (1) deal with physical and emotional stress, and (2) maintain adequate energy supply and blood sugar levels. Particularly, the adrenal glands are controlled by the pituitary gland.
The pituitary gland is a small pea-sized gland at the base of the brain. In the event that the pituitary gland senses that there is not enough cortisol present in the bloodstream, it releases a hormone called ACTH (adrenocorticotropic hormone). Then, ACTH stimulates the adrenals to produce more cortisol. However, those with CAH have insufficient amounts of the enzyme 21-hydroxylase, needed to convert a precursor molecule called 17-hydroxyprogesterone (17-OHP) into cortisol.
As a result, the pituitary gland continues to sense the need for cortisol and pumps out more ACTH. This leads to an overabundance of 17-OHP, which is converted in the adrenals into excess androgens (masculinizing steroid hormones) (see figure below left). Lack of adequate cortisol also prevents the body from properly metabolizing sugar and responding to stress. The lack of this stress response can lead to an adrenal crisis.
Classic CAH
Moreover, greater than 75% of all individuals with Classic CAH also lack another adrenal hormone called aldosterone, necessary for maintaining normal fluid volume of the body, sodium and potassium, which among other functions, stabilizes the heart. When this deficiency occurs it is called “Salt-Wasting CAH” (SW-CAH). The remaining 25 percent of those with Classical CAH who produce sufficient aldosterone are referred to as “Simple Virilizers” (SV-CAH).
Non-Classic CAH
Furthermore, the non-classic form of CAH is not life threatening but can affect: (1) puberty and growth in children, and (2) cause infertility in males and females as well as other symptoms affecting quality of life.
Additionally, other rare forms of enzyme deficiency that belong to the Congenital Adrenal Hyperplasia family of disorders are: 3Beta-hydroxysteroid dehydrogenase deficiency (3B-HSD), 11-Beta hydroxylase deficiency (11B-HD) and 17-alpha hydroxylase deficiency (17a-HD) which are much less common. This website focuses on 21-OH deficiency, as it is the most common form of the disorder.

Click the icons below to learn more!
Join the CARES Community (free to join)
We encourage all of those who have a connection to congenital adrenal hyperplasia (CAH) to join the CAH Community with CARES Foundation. Furthermore, joining the CARES community is completely free, and the best way to stay up to date on important information, updates, events, research, and more!
You can create your free account with CARES Foundation by visiting: https://caresfoundation.org/join-the-cares-community/.
Resources
I’m Growing with CAH! A book for children about CAH. Featured on the Minnesota Health Dept website
Cresendo Com HAC! Portuguese (Brazilian) version
CAH Booklet for Parents/Caregivers from British Columbia’s Children’s Hospital

CONTACT
2414 Morris Avenue, Ste 110
Union, NJ 07083
Phone: (908) 364-0272
Toll Free: (866) 227-3737
Fax: (908) 686-2019
contact@caresfoundation.org
